
Welcome to our tutorial on GPU-accelerated AMBER! We make it easy to benchmark your applications and problem sets on the latest hardware. Our GPU Test Drive Cluster provides developers, scientists, academics, and anyone else interested in GPU computing with the opportunity to test their code. While Test Drive users are given free reign to use their own applications on the cluster, Microway also provides a variety of pre-installed GPU accelerated applications.
In this post, we will look at the molecular dynamics package AMBER. Collaboratively developed by professors at a variety of university labs, the latest versions of AMBER natively support GPU acceleration. We’ll demonstrate how NVIDIA® Tesla® K40 GPUs can deliver a speedup of up to 86X!
Before we jump in, we should mention that this post assumes you are familiar with AMBER and/or AmberTools. If you are more familiar with another molecular dynamics package (e.g., GROMACS), check to see what we already have pre-installed on our cluster. There may be no need for you to learn a new tool. If you’re new to these tools in general, you can find quite a large number of AMBER tutorials online.
Access our GPU-accelerated Test Cluster
Getting access to the Microway Test Drive cluster is fast and easy – fill out a short form to sign up for a GPU Test Drive. Although our approval e-mail includes a list of commands to help you get your benchmark running, we’ll go over the steps in more detail below.
First, you need to log in to the Microway Test Drive cluster using SSH. Don’t worry if you’re unfamiliar with SSH – we include a step-by-step instruction manual for logging in. SSH is built-in on Linux and MacOS; Windows users need to install one application.
Run CPU and GPU versions of AMBER
This is one of the easiest steps. Just enter the AMBER directory and run the default benchmark script which we have pre-written for you:
cd amber sbatch run-amber-on-TeslaK40.sh
Waiting for jobs to complete
Our cluster uses SLURM for resource management. Keeping track of your job is easy using the squeue command. For real-time information on your job, run: watch squeue (hit CTRL+c to exit). Alternatively, you can tell the cluster to e-mail you when your job is finished by editing the AMBER batch script file (although this must be done before submitting jobs with sbatch). Run:
nano run-amber-on-TeslaK40.sh
Within this file, add the following two lines to the #SBATCH section (specifying your own e-mail address):
#SBATCH --mail-user=[email protected] #SBATCH --mail-type=END
If you would like to monitor the compute node which is running your job, examine the output of squeue and take note of which node your job is running on. Log into that node using SSH and then use the tools of your choice to monitor it. For example:
ssh node2 nvidia-smi htop
(hit q to exit htop)
See the speedup of GPUs vs. CPUs
The results from our benchmark script will be placed in an output file called amber-K40.xxxx.output.log – below is a sample of the output running on CPUs:
=============================================================== = Run CPU-only: JAC_PRODUCTION_NVE - 23,558 atoms PME =============================================================== | ns/day = 25.95 seconds/ns = 3329.90
and with AMBER running on GPUs (demonstrating a 6X speed-up):
======================================================================== = Run Tesla_K40m GPU-accelerated: JAC_PRODUCTION_NVE - 23,558 atoms PME ======================================================================== | ns/day = 157.24 seconds/ns = 549.47
Should you require more information on a particular run, it’s available in the benchmarks/ directory (with a separate subdirectory for each test case). If your job has any problems, the errors will be logged to the file amber-K40.xxxx.output.errors
The chart below demonstrates the performance improvements between a CPU-only AMBER run (on two 10-core Ivy Bridge Intel Xeon CPUs) and a GPU-accelerated AMBER run (on two NVIDIA Tesla K40 GPUs):
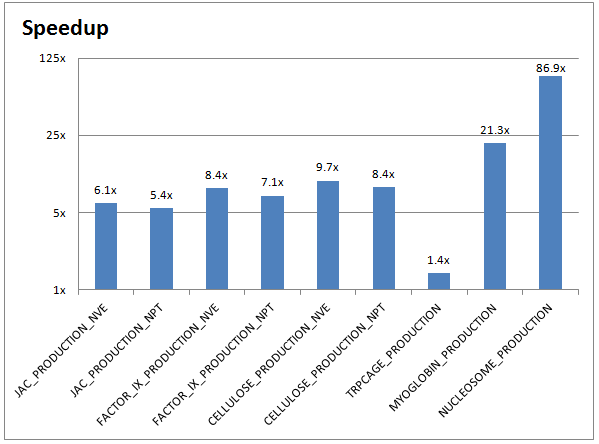
Running your own AMBER inputs on GPUs
If you’re familiar with BASH, you can of course create your own batch script, but we recommend using the run-amber-your-files.sh file as a template for when you want to run you own simulations. For AMBER, the key files are the prmtop, inpcrd, and mdin files. You can upload these files yourself or you can build them. If you opt for the latter, you need to load the appropriate software packages by running:
module load cuda gcc mvapich2-cuda amber
Once your files are either created or uploaded, you’ll need to ensure that the batch script is referencing the correct input files. The relevant parts of the run-amber-your-files.sh file are:
echo "===============================================================" echo "= Run CPU-only" echo "===============================================================" srun -n $NPROCS pmemd.MPI -O -i mdin -o mdout.cpu -p prmtop -inf mdinfo.cpu -c inpcrd -r restrt.cpu -x mdcrd.cpu grep "ns/day" mdinfo.cpu | tail -n1
and for execution on GPUs:
echo "===============================================================" echo "= Run GPU-Accelerated" echo "===============================================================" srun -n $GPUS_PER_NODE pmemd.cuda.MPI -O -i mdin -o mdout.cpu -p prmtop -inf mdinfo.gpu -c inpcrd -r restrt.gpu -x mdcrd.gpu grep "ns/day" mdinfo.gpu | tail -n1
The above script assumes that mdin (control data: variables and simulation options), prmtop (topology: the molecular topology and force field parameters), and inpcrd (coordinates: the atom coordinates, velocities, box dimensions) are the main input files, but you are free to add additional levels of complexity as well. The output files (mdout, mdinfo, restrt, mdcrd) are labeled with the suffixes .cpu and .gpu. The line which lists the grep command is used to populate the amber-K40.xxxx.output.log output file with the ns/day benchmark times (just as shown in the sample output listed above).
If you’d like to visualize your results, you will need an SSH client which properly forwards your X-session. You are welcome to contact us if you’re uncertain of this step. Once that’s done, the VMD visualization tool can be accessed by running:
module load vmd vmd
What’s next?
With the right setup (which we’ve already done for you), running AMBER on a GPU cluster isn’t much more difficult than running it on your own workstation. We also make it easy to compare benchmark results between CPUs and GPUs. If you’d like to learn more, contact one of our experts or sign up for a GPU Test Drive today!
Citations for AMBER and AmberTools:
D.A. Case, T.A. Darden, T.E. Cheatham, III, C.L. Simmerling, J. Wang, R.E. Duke, R. Luo, R.C. Walker, W. Zhang, K.M. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A.W. Goetz, I. Kolossváry, K.F. Wong, F. Paesani, J. Vanicek, R.M. Wolf, J. Liu, X. Wu, S.R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D.R. Roe, D.H. Mathews, M.G. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko, and P.A. Kollman (2012), AMBER 12, University of California, San Francisco.
PME: Romelia Salomon-Ferrer; Andreas W. Goetz; Duncan Poole; Scott Le Grand; & Ross C. Walker* “Routine microsecond molecular dynamics simulations with AMBER – Part II: Particle Mesh Ewald” , J. Chem. Theory Comput., 2013, 9 (9), pp 3878-3888, DOI: 10.1021/ct400314y
GB: Andreas W. Goetz; Mark J. Williamson; Dong Xu; Duncan Poole; Scott Le Grand; & Ross C. Walker* “Routine microsecond molecular dynamics simulations with AMBER – Part I: Generalized Born”, J. Chem. Theory Comput., (2012), 8 (5), pp 1542-1555, DOI: 10.1021/ct200909j
Citation for VMD:
Humphrey, W., Dalke, A. and Schulten, K., “VMD – Visual Molecular Dynamics” J. Molec. Graphics 1996, 14.1, 33-38
