This is a guest post by Adam Marko, an IT and Biotech Professional with 10+ years of experience across diverse industries
What is Bowtie2?
Bowtie2 is a commonly used, open-source, fast, and memory efficient application used as part of a Next Generation Sequencing (NGS) workflow.It aligns the sequencing reads, which are the genomic data output from an NGS device such as an Illumina HiSeq Sequencer, to a reference genome.Applications like Bowtie2 are used as the first step in pipelines such as those for variant determination, and an area of continuously growing research interest, RNA-Seq.
What is RNA-Seq?
RNA Sequencing (RNA-Seq) is a type of NGS that seeks to identify the presence and quantity of RNA in a sample at a given point in time.This can be used to quantify changes in gene expression, which can be a result of time, external stimuli, healthy or diseased states, and other factors.Through this quantification, researchers can obtain a unique snapshot of the genomic status of the organism to identify genomic information previously undetectable with other technologies.
There is considerable research effort being put into RNA-Seq, and the number of publications has grown steadily since its first use in 2009.
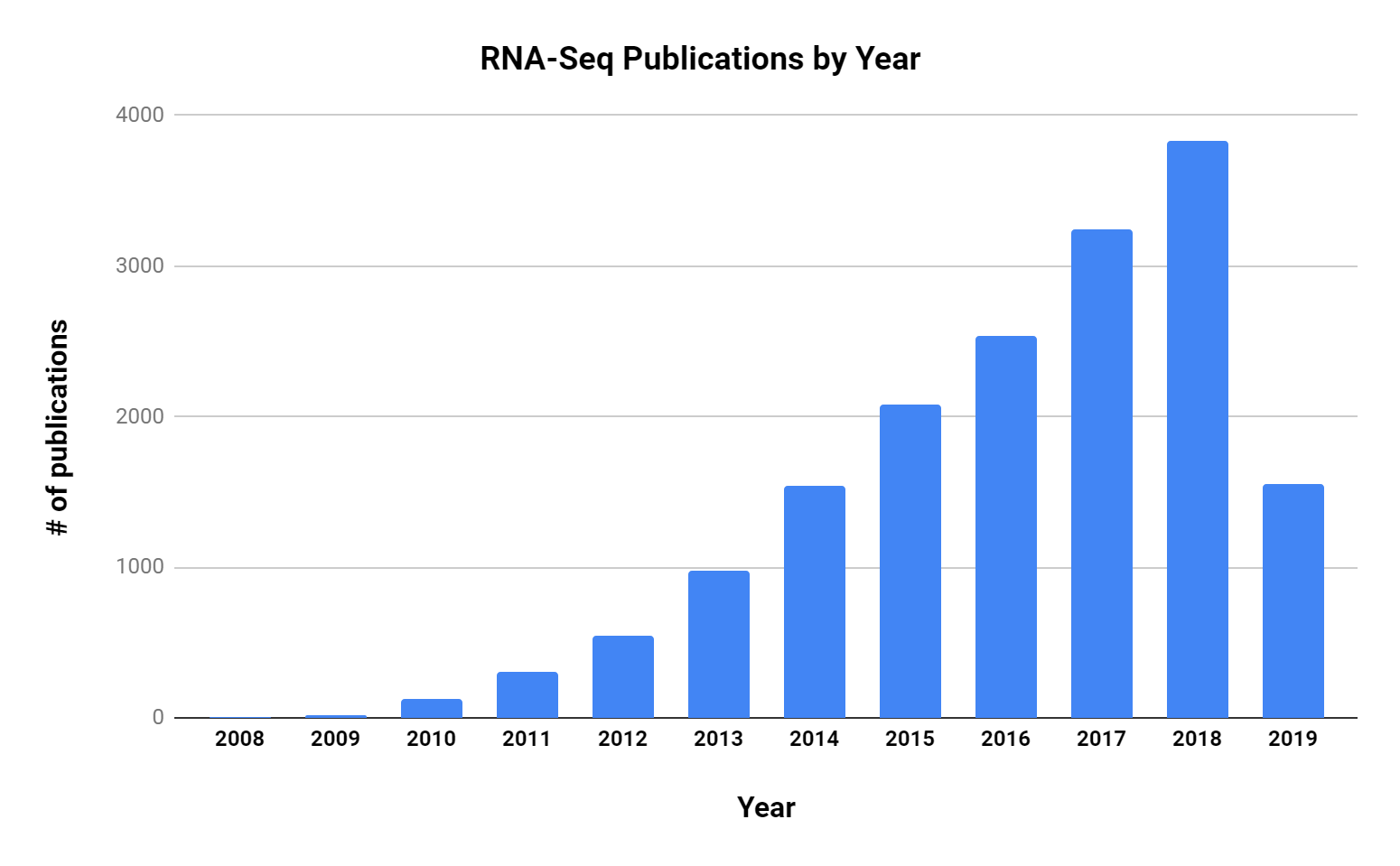
RNA-Seq is being applied to many research areas and diseases, and a few notable examples of using the technology include:
- Oral Cancer: Researchers used an RNA-Seq approach to identify differences in gene expression between oral cancer and normal tissue samples.
- Alzheimer’s Disease: Researchers compared the gene expression of different lobes of deceased Alzheimer’s Disease patients brain with the brain of healthy individuals.They were able to identify genomic differences between the diseased and unaffected individuals.
- Diabetes: Researchers identified novel gene expression information from pancreatic beta-cells, which are cells critical for glycemic control.
Compute Infrastructure for aligning with Bowtie2
Designing a compute resource to meet the sequence analysis needs of Bioinformatics researchers can be a daunting task for IT staff.Limited information is available about multithreading and performance increases in the diverse portfolio of software related to NGS analysis.To further complicate things, processors are now available in a variety of models, with a large range of core counts and clock speeds, from both AMD and Intel. See, for example, the latest Intel Xeon “Cascade Lake” CPUs: Intel Xeon Scalable “Cascade Lake SP” Processor Review
Though many sequence analysis tools have multithreading options, the ability to scale is often limited, and rarely linear.In some cases, performance can decrease as more threads are added.Multithreading applications does not guarantee a performance improvement.
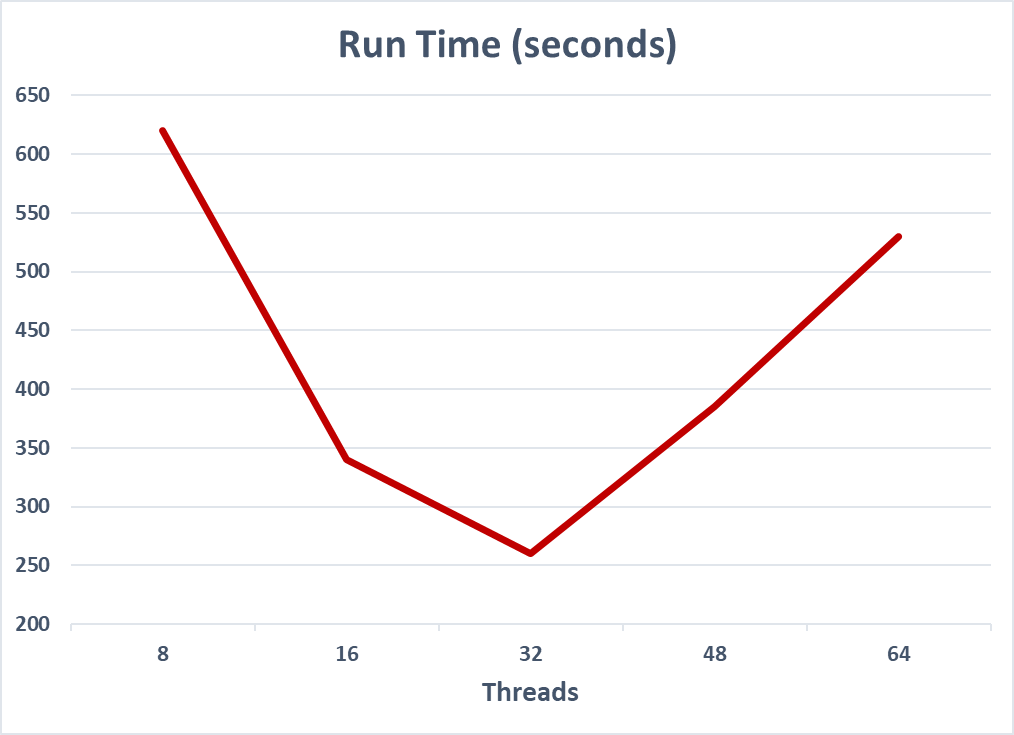
| Threads | Run Time (seconds) |
|---|---|
| 8 | 620 |
| 16 | 340 |
| 32 | 260 |
| 48 | 385 |
| 64 | 530 |
Table 1. Research data showing previous version of Bowtie2 scaling with thread count.Performance would decrease above 32 threads.
However, researchers recently greatly improved the thread scaling of Bowtie2.Original versions of this tool did not scale linearly, and demonstrated reduced performance when using more than 32 threads.Aware of these problems, the developers of Bowtie2 have implemented superior multithread scaling in their applications.Depending on processor type, their results show:
- Removal of performance decreases over 32 threads
- An increase in read throughput of up to 44%
- Reduced memory usage with thread scaling
- Up to a 4 hour reduction in time to align 40x coverage human genome
This new version of the software is open-source and available for download.
Right Sizing your NGS Cluster
With the recent release of Intel’s Cascade Lake-AP Xeons providing up to 112 threads per socket, as well as high density AMD EPYC processors, it can be tempting to assume that more cores will result in more performance for NGS applications.However, this is not always the case, and some applications will show reduced performance with higher thread count.
When selecting compute systems for NGS analysis, researchers and IT staff need to evaluate which software products will be used, and how they scale with threads.Depending on the use cases, more nodes with fewer, faster, threads could provide better performance than high thread density nodes.Unfortunately there is no “one size fits all” solution, and applications are in constant development, so research into the most recent versions of analysis software is always required.
References
[1] https://www.ncbi.nlm.nih.gov/pubmed/
[2] https://doi.org/10.1371/journal.pone.0016266
[3] https://doi.org/10.1101/205328
[4] https://link.springer.com/article/10.1007/s10586-017-1015-0
