A powerful new open source deep learning framework for drug discovery is now available for public download on github.This new framework, called DeepChem, is python-based, and offers a feature-rich set of functionality for applying deep learning to problems in drug discovery and cheminformatics.Previous deep learning frameworks, such as scikit-learn have been applied to chemiformatics, but DeepChem is the first to accelerate computation with NVIDIA GPUs.
The framework uses Google TensorFlow, along with scikit-learn, for expressing neural networks for deep learning.It also makes use of the RDKit python framework, for performing more basic operations on molecular data, such as converting SMILES strings into molecular graphs.The framework is now in the alpha stage, at version 0.1.As the framework develops, it will move toward implementing more models in TensorFlow, which use GPUs for training and inference.This new open source framework is poised to become an accelerating factor for innovation in drug discovery across industry and academia.
Another unique aspect of DeepChem is that it has incorporated a large amount of publicly-available chemical assay datasets, which are described in Table 1.
DeepChem Assay Datasets
| Dataset | Category | Description | Classification Type | Compounds |
|---|---|---|---|---|
| QM7 | Quantum Mechanics | orbital energies atomization energies | Regression | 7,165 |
| QM7b | Quantum Mechanics | orbital energies | Regression | 7,211 |
| ESOL | Physical Chemistry | solubility | Regression | 1,128 |
| FreeSolv | Physical Chemistry | solvation energy | Regression | 643 |
| PCBA | Biophysics | bioactivity | Classification | 439,863 |
| MUV | Biophysics | bioactivity | Classification | 93,127 |
| HIV | Biophysics | bioactivity | Classification | 41,913 |
| PDBBind | Biophysics | binding activity | Regression | 11,908 |
| Tox21 | Physiology | toxicity | Classification | 8,014 |
| ToxCast | Physiology | toxicity | Classification | 8,615 |
| SIDER | Physiology | side reactions | Classification | 1,427 |
| ClinTox | Physiology | clinical toxicity | Classification | 1,491 |
Table 1:The current v0.1 DeepChem Framework includes the data sets in this table, along others which will be added to future versions.
Metrics
The squared Pearson Correleation Coefficient is used to quantify the quality of performance of a model trained on any of these regression datasets.Models trained on classification datasets have their predictive quality measured by the area under curve (AUC) for receiver operator characteristic (ROC) curves (AUC-ROC).Some datasets have more than one task, in which case the mean over all tasks is reported by the framework.
Data Splitting
DeepChem uses a number of methods for randomizing or reordering datasets so that models can be trained on sets which are more thoroughly randomized, in both the training and validation sets, for example.These methods are summarized in Table 2.
DeepChem Dataset Splitting Methods
| Split Type | use cases |
|---|---|
| Index Split | default index is sufficient as long as it contains no built-in bias |
| Random Split | if there is some bias to the default index |
| Scaffold Split | if chemical properties of dataset will be depend on molecular scaffold |
| Stratified Random Split | where one needs to ensure that each dataset split contains a full range of some real-valued property |
Table 2:Various methods are available for splitting the dataset in order to avoid sampling bias.
Featurizations
DeepChem offers a number of featurization methods, summarized in Table 3.SMILES strings are unique representations of molecules, and can themselves can be used as a molecular feature.The use of SMILES strings has been explored in recent work.SMILES featurization will likely become a part of future versions of DeepChem.
Most machine learning methods, however, require more feature information than can be extracted from a SMILES string alone.
DeepChem Featurizers
| Featurizer | use cases |
|---|---|
| Extended-Connectivity Fingerprints (ECFP) | for molecular datasets not containing large numbers of non-bonded interactions |
| Graph Convolutions | Like ECFP, graph convolution produces granular representations of molecular topology. Instead of applying fixed hash functions, as with ECFP, graph convolution uses a set of parameters which can learned by training a neural network associated with a molecular graph structure. |
| Coloumb Matrix | Coloumb matrix featurization captures information about the nuclear charge state, and internuclear electric repulsion. This featurization is less granular than ECFP, or graph convolutions, and may perform better where intramolecular electrical potential may play an important role in chemical activity |
| Grid Featurization | datasets containing molecules interacting through non-bonded forces, such as docked protein-ligand complexes |
Table 3:Various methods are available for splitting the dataset in order to avoid sampling bias.
Supported Models
Supported Models as of v0.1
| Model Type | possible use case |
|---|---|
| Logistic Regression | continuous, real-valued prediction required |
| Random Forest | Classification or Regression |
| Multitask Network | If various prediction types required, a multitask network would be a good choice. An example would be a continuous real-valued prediction, along with one or more categorical predictions, as predicted outcomes. |
| Bypass Network | Classification and Regression |
| Graph Convolution Model | same as Multitask Networks |
Table 4: Model types supported by DeepChem 0.1
A Glimpse into the Tox21 Dataset and Deep Learning
The Toxicology in the 21st Century (Tox21) research initiative led to the creation of a public dataset which includes measurements of activation of stress response and nuclear receptor response pathways by 8,014 distinct molecules.Twelve response pathways were observed in total, with each having some association with toxicity.Table 5 summarizes the pathways investigated in the study.
Tox21 Assay Descriptions
| Biological Assay | description |
|---|---|
| NR-AR | Nuclear Receptor Panel, Androgen Receptor |
| NR-AR-LBD | Nuclear Receptor Panel, Androgen Receptor, luciferase |
| NR-AhR | Nuclear Receptor Panel, aryl hydrocarbon receptor |
| NR-Aromatase | Nuclear Receptor Panel, aromatase |
| NR-ER | Nuclear Receptor Panel, Estrogen Receptor alpha |
| NR-ER-LBD | Nuclear Receptor Panel, Estrogen Receptor alpha, luciferase |
| NR-PPAR-gamma | Nuclear Receptor Panel, peroxisome profilerator-activated receptor gamma |
| SR-ARE | Stress Response Panel, nuclear factor (erythroid-derived 2)-like 2 antioxidant responsive element |
| SR-ATAD5 | Stress Response Panel, genotoxicity indicated by ATAD5 |
| SR-HSE | Stress Response Panel, heat shock factor response element |
| SR-MMP | Stress Response Panel, mitochondrial membrane potential |
| SR-p53 | Stress Response Panel, DNA damage p53 pathway |
Table 5:Biological pathway responses investigated in the Tox21 Machine Learning Challenge.
We used the Tox21 dataset to make predictions on molecular toxicity in DeepChem using the variations shown in Table 6.
Model Construction Parameter Variations Used
| Dataset Splitting | Index | Scaffold |
| Featurization | ECFP | Molecular Graph Convolution |
Table 6:Model construction parameter variations used in generating our predictions, as shown in Figure 1.
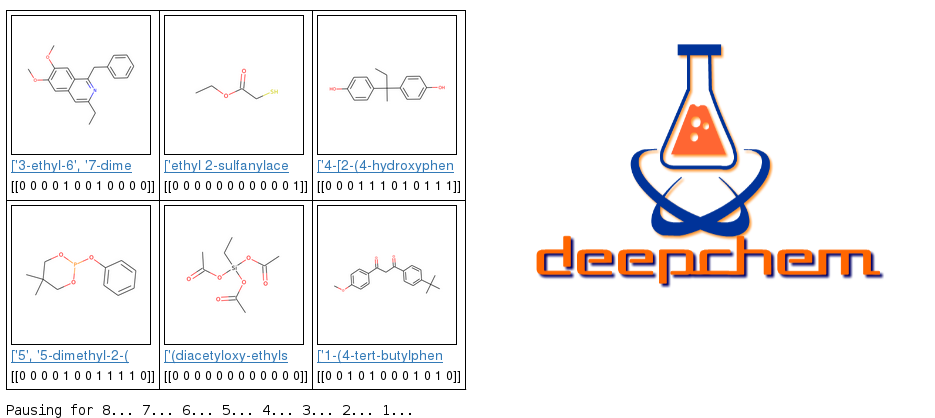
A .csv file containing SMILES strings for 8,014 molecules was used to first featurize each molecule by using either ECFP or molecular graph convolution.IUPAC names for each molecule were queried from NIH Cactus, and toxicity predictions were made, using a trained model, on a set of nine molecules randomly selected from the total tox21 data set.Nine results showing molecular structure (rendered by RDKit), IUPAC names, and predicted toxicity scores, across all 12 biochemical response pathways, described in Table 5, are shown in Figure 1.

Expect more from DeepChem in the Future
The DeepChem framework is undergoing rapid development, and is currently at the 0.1 release version.New models and features will be added, along with more data sets in future.You can download the DeepChem framework from github.There is also a website for framework documentation at deepchem.io.
Microway offers DeepChem pre-installed on our line of WhisperStation products for Deep Learning. Researchers interested in exploring deep learning applications with chemistry and drug discovery can browse our line of WhisperStation products.
References
1.) Subramanian, Govindan, et al. “Computational Modeling of β-secretase 1 (BACE-1) Inhibitors using Ligand Based Approaches.” Journal of Chemical Information and Modeling 56.10 (2016): 1936-1949.
2.) Altae-Tran, Han, et al. “Low Data Drug Discovery with One-shot Learning.” arXiv preprint arXiv:1611.03199 (2016).
3.) Wu, Zhenqin, et al. “MoleculeNet: A Benchmark for Molecular Machine Learning.” arXiv preprint arXiv:1703.00564 (2017).
4.) Gomes, Joseph, et al. “Atomic Convolutional Networks for Predicting Protein-Ligand Binding Affinity.” arXiv preprint arXiv:1703.10603 (2017).
5.) Gómez-Bombarelli, Rafael, et al. “Automatic chemical design using a data-driven continuous representation of molecules.” arXiv preprint arXiv:1610.02415 (2016).
6.) Mayr, Andreas, et al. “DeepTox: toxicity prediction using deep learning.” Frontiers in Environmental Science 3 (2016): 80.
