This is a guest post by Adam Marko, an IT and Biotech Professional with 10+ years of experience across diverse industries
Background and history
Cryogenic Electron Microscopy (CryoEM) is a type of electron microscopy that images molecular samples embedded in a thin layer of non-crystalline ice, also called vitreous ice.Though CryoEM experiments have been performed since the 1980s, the majority of molecular structures have been determined with two other techniques, X-ray crystallography and Nuclear Magnetic Resonance (NMR).The primary advantage of X-ray crystallography and NMR is that molecules were able to be determined at very high resolution, several fold better than historical CryoEM results.
However, recent advancements in CryoEM microscope detector technology and analysis software have greatly improved the capability of this technique.Before 2012, CryoEM structures could not achieve the resolution of X-ray Crystallography and NMR structures.The imaging and analysis improvements since that time now allow researchers to image structures of large molecules and complexes at high resolution.The primary advantages of Cryo-EM over X-ray Crystallography and NMR are:
- Much larger structures can be determined than by X-ray or NMR
- Structures can be determined in a more native state than by using X-ray
The ability to generate these high resolution large molecular structures through CryoEM enables better understanding of life science processes and improved opportunities for drug design. CryoEM has been considered so impactful, that the inventors won the 2017 Nobel Prize in chemistry.
CryoEM structure and publication growth
While the number of molecular structures determined by CryoEM is much lower than those determined by X-ray crystallography and NMR, the rate at which these structures are released has greatly increased in the past decade. In 2016, the number CryoEM structures deposited in the Protein Data Bank (PDB) exceeded those of NMR for the first time.
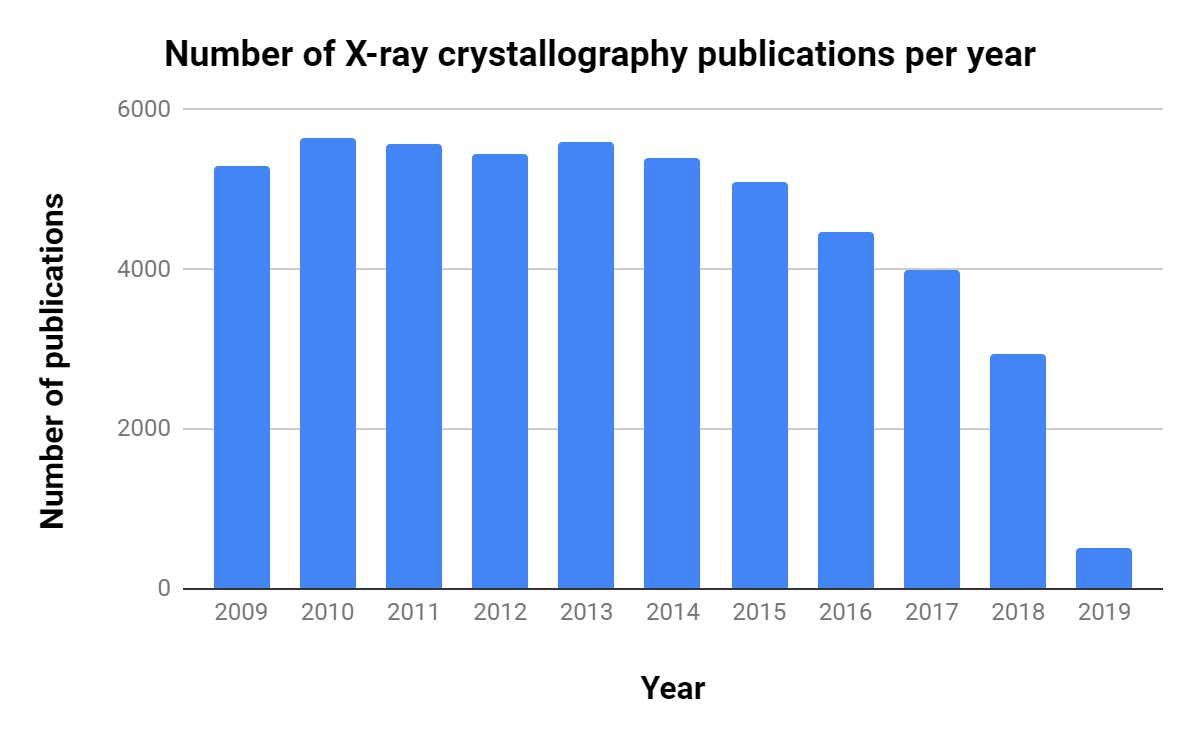
The importance of CryoEM in research is growing, as shown by the steady increase in publications over the past decade (Table 1, Figure 1).Interestingly, though there are ~10 times as many X-ray crystallography publications per year, the number of related publications per year has decreased consistently since 2013.
| Experimental Structure Type | Approximate total number of structures as of March 2019 |
|---|---|
| X-ray Crystallography | 134,000 |
| NMR | 12,500 |
| Cryo-EM | 3,000 |
Table 1. Total number of CryoEM structures available in the publicly accessible Protein Data Bank (PDB). Source: RCSB
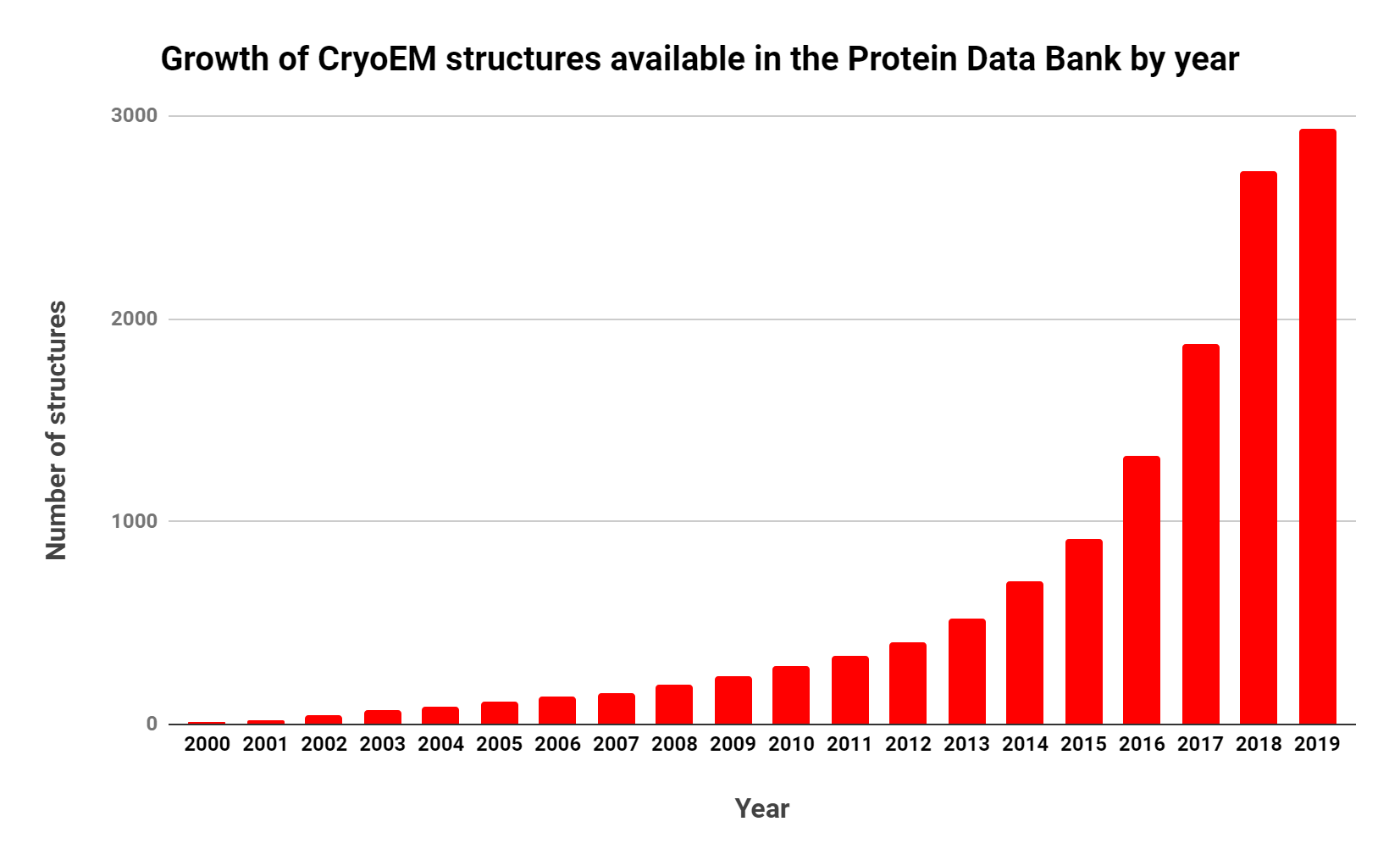
Note the rapid and consistent growth. Source: RCSB
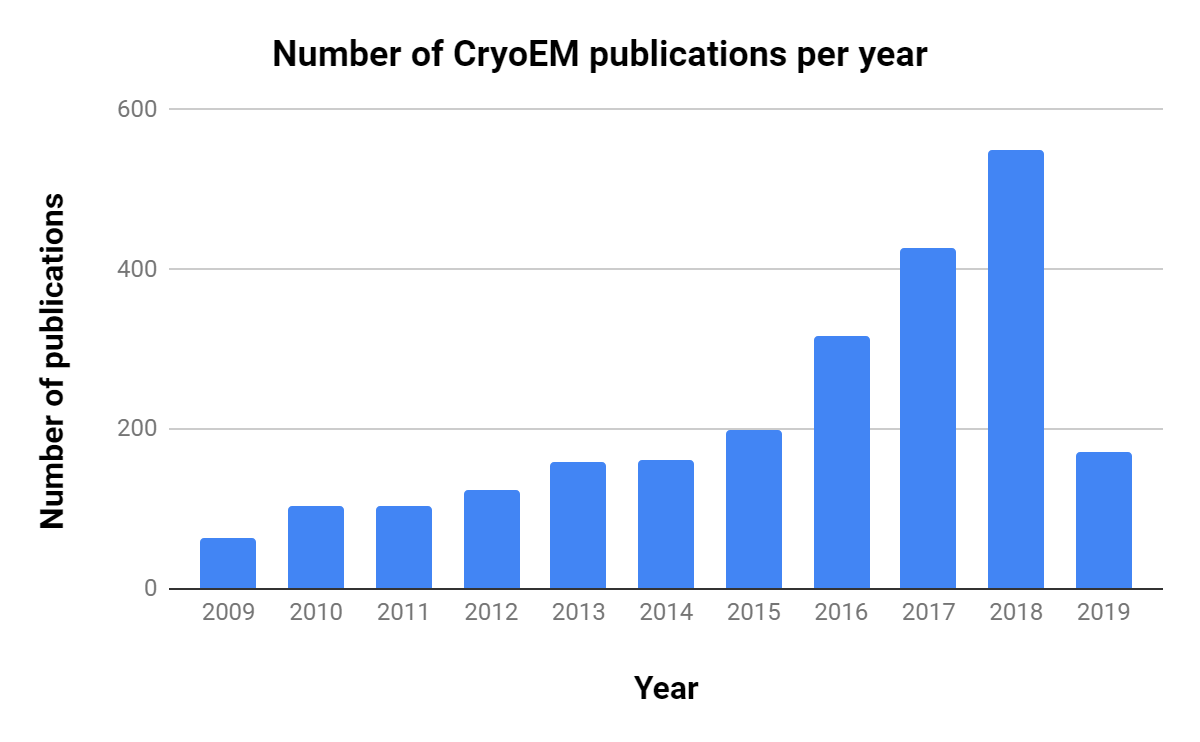
Note the rapid increase in publications. Source: NCBI PubMed
CryoEM is part of our research – do I need to add GPUs to my infrastructure?
A major challenge facing researchers and IT staff is how to appropriately build out infrastructure for CryoEM demands.There are several software products that are used for CryoEM analysis, with RELION being one of the most widely used open source packages.While GPUs can greatly accelerate RELION workflows, support for them has only existed since Version 2 (released in 2016).Worldwide, the vast majority of individual servers and centralized resources available to researchers are not GPU accelerated.Those systems that do have professional grade GPUs are often oversubscribed and can have considerable queue wait times.The relative high cost of server grade GPU systems can put those devices out of the reach of many individual research labs.
While advanced GPU hardware like the DGX-1 continue to give the best analysis times, not every GPU system provides the same throughput. Large datasets can create issues with consumer grade GPUs, in that the dataset must fit within the GPU memory to fully take advantage of the acceleration.Though RELION can parallelize the datasets, GPU memory is still limited when compared to the large amounts of system memory available to CPUs that can be installed in a single device (DGX-1 provides 256GB GPU memory; DGX-2 provides 512GB).This problem is amplified if the researcher has access to only a single consumer grade graphic card (e.g., an NVIDIA GeForce GTX 1080 Ti GPU with 11GB memory).
With the Version 3 release of the software (late 2018), RELION authors have implemented CPU acceleration to broaden the usable hardware for efficient CryoEM reconstruction.The authors have shown a 1.5x improvement on Broadwell processors and a 2.5x improvement on Skylake over the previous code.However, taking advantage of AVX instructions during compilation can further improve performance, with the authors demonstrating a 5.4x improvement on Skylake processors.This improvement is approaching the performance increases of professional grade GPUs without the additional cost.
Additional infrastructure considerations
CryoEM datasets are being generated at a higher rate and with larger data sizes than ever before.Currently, the largest raw dataset in the Electron Microscopy Public Image Archive (EMPIAR) is 12.4TB, with a median dataset size of approximately 2TB.Researchers and IT staff can expect datasets in this order of magnitude to become the norm as CryoEM continues to grow as an experimental resource in the life sciences space.
Many CryoEM labs function as microscopy cores, where they provide the service of generating the 2D datasets for different researchers, which are then analyzed by individual labs.Given the high cost of professional GPUs as compared to the ubiquitous availability of multicore CPU systems, researchers may consider modern multicore servers or using centralized clusters to meet their CryoEM analysis needs.This is with the caveat that they use Version 3 of RELION software with appropriate compilation flags.
Dataset transfer is also a concern, and organizations that have a centralized Cryo-EM core would greatly benefit from upgraded networking (10Gbps+) from the core location to centralized compute resources, or to individual labs.
CryoEM takes center stage
The increase in capabilities, interest, and research related to CryoEM shows it is now a mainstream experimental technique.IT staff and scientists alike are rapidly becoming aware of this fact as they face the data analysis, transfer, and storage challenges associated with this technique.Careful consideration must be given to the infrastructure of an organization that is engaging in CryoEM research.
In an organization that is performing exclusively CryoEM experiments, a GPU cluster would be the most cost-effective solution for rapid analysis.Researchers with access to advanced professional grade GPU systems, such as a DGX-1, will see analysis times that are even faster than modern CPU optimized RELION.While these professional GPUs can greatly accelerate CryoEM analysis, it is unlikely in the short term that all researchers wanting to use CryoEM data will have access to such high-spec GPU hardware, as compared to mixed-use commodity clusters, which are ubiquitous at all life science organizations.A large multicore CPU machine, when properly configured, can give better performance than a low core workstation or server with a single consumer grade GPU (e.g., an NVIDIA GeForce GPU).
IT departments and researchers must work together to define expected turnaround time, analysis workflow requirements, budget, and configuration of existing hardware.In doing so, researcher needs will be met and IT can implement the most effective architecture for CryoEM.
References
[1] https://doi.org/10.7554/eLife.42166.001
[2] https://febs.onlinelibrary.wiley.com/doi/10.1111/febs.12796
[3] https://www.ncbi.nlm.nih.gov/pubmed/
[4] https://www.ebi.ac.uk/pdbe/emdb/empiar/
[5] https://www.rcsb.org/
