This is a tutorial on the usage of GPU-accelerated NAMD for molecular dynamics simulations. We make it simple to test your codes on the latest high-performance systems – you are free to use your own applications on our cluster and we also provide a variety of pre-installed applications with built-in GPU support. Our GPU Test Drive Cluster acts as a useful resource for demonstrating the increased application performance which can be achieved with NVIDIA Tesla GPUs.
This post describes the scalable molecular dynamics software NAMD, which comes out of the Theoretical and Computational Biophysics Group at the University of Illinois Urbana-Champaign. NAMD supports a variety of operational modes, including GPU-accelerated runs across large numbers of compute nodes. We’ll demonstrate how a single server with NVIDIA® Tesla® K40 GPUs can deliver speedups over 4X!
Before continuing, please note that this post assumes you are familiar with NAMD. If you prefer a different molecular dynamics package (e.g., AMBER), read through the list of applications we have pre-installed. There may be no need for you to learn a new tool. If all of these tools are new to you, you will find a number of NAMD tutorials online.
Access the Tesla GPU-accelerated Cluster
Getting started with our GPU Benchmark cluster is fast and easy – fill out this short form to sign up for GPU benchmarking. Although we will send you an e-mail with a general list of commands when your request is accepted, this post goes into further detail.
First, you need to log in to the GPU cluster using SSH. Don’t worry if you haven’t used SSH before – we will send you step-by-step login instructions. Windows users have to perform one additional step, but SSH is built-in on Linux and MacOS.
Run CPU and GPU-accelerated versions of NAMD
Once you’re logged in, it’s easy to compare CPU and GPU performance: enter the NAMD directory and run the NAMD batch script which we have pre-written for you:
cd namd sbatch run-namd-on-TeslaK40.sh
Waiting for your NAMD job to finish
Our cluster uses SLURM to manage users’ jobs. You can use the squeue command to keep track of your jobs. For real-time information on your job, run: watch squeue (hit CTRL+c to exit). Alternatively, the cluster can e-mail you when your job is finished if you update the NAMD batch script file (although this must be done before submitting your job). Run:
nano run-namd-on-TeslaK40.sh
Within this file, add the following two lines to the #SBATCH section (changing the e-mail address to your own):
#SBATCH --mail-user=[email protected] #SBATCH --mail-type=END
If you would like to closely monitor the compute node which is running your job, check the output of squeue and take note of which compute node your job is running on. Log into that node with SSH and then use one of the following tools to keep an eye on GPU and system status:
ssh node2 nvidia-smi htop
(hit q to exit htop)
Check the speedup of NAMD on GPUs vs. CPUs
The results from the NAMD batch script will be placed in an output file named namd-K40.xxxx.output.log – below is a sample of the output running on CPUs:
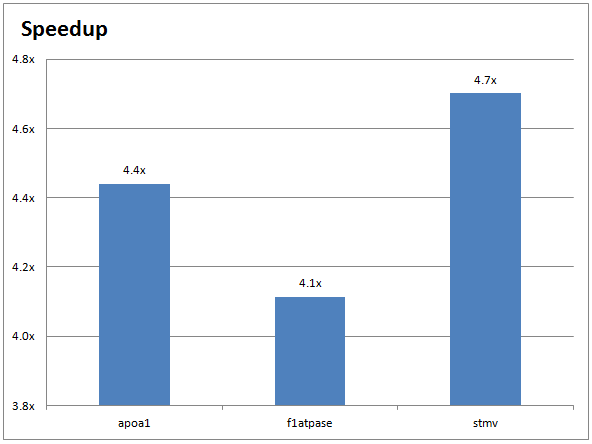
====================================================== = Run CPU only stmv ====================================================== Info: Benchmark time: 20 CPUs 0.531318 s/step 6.14951 days/ns 4769.63 MB memory
and with NAMD running on two GPUs (demonstrating over 4X speed-up):
====================================================== = Run Tesla_K40m GPU-accelerated stmv ====================================================== Info: Benchmark time: 18 CPUs 0.112677 s/step 1.30413 days/ns 2475.9 MB memory
Should you require further details on a particular run, you will see that a separate log file has been created for each of the inputs (e.g., stmv.20_cpu_cores.output). The NAMD output files are available in the benchmarks/ directory (with a separate subdirectory for each test case). If your job has any problems, the errors will be logged to the file namd-K40.xxxx.output.errors
The following chart shows the performance improvements for a CPU-only NAMD run (on two 10-core Ivy Bridge Intel Xeon CPUs) versus a GPU-accelerated NAMD run (on two NVIDIA Tesla K40 GPUs):
Running your own NAMD inputs on GPUs
If you’re familiar with BASH you can write your own batch script from scratch, but we recommend using the run-namd-your-files.sh file as a template when you’d like to try your own simulations. For most NAMD runs, the batch script will only reference a single input file (e.g., the stmv.namd script). This input script will reference any other input files which NAMD might require:
- Structure file (e.g.,
stmv.psf) - Coordinates file (e.g.,
stmv.pdb) - Input parameters file (e.g.,
par_all27_prot_na.inp)
You can upload existing inputs from your own workstation/laptop or you can assemble an input job on the cluster. If you opt for the latter, you need to load the appropriate software packages by running:
module load cuda gcc namd
Once your files are in place in your namd/ directory, you’ll need to ensure that the batch script is referencing the correct .namd input file. The relevant lines of the run-namd-your-files.sh file are:
echo "===============================================================" echo "= Run CPU-only" echo "===============================================================" namd2 +p $num_cores_cpu input_file.namd > namd_output__cpu_run.txt grep Benchmark namd_output__cpu_run.txt
and for execution on GPUs:
echo "===============================================================" echo "= Run GPU-Accelerated" echo "===============================================================" namd2 +p $num_cores_gpu +devices $CUDA_VISIBLE_DEVICES +idlepoll input_file.namd > namd_output__gpu_run.txt grep Benchmark namd_output__gpu_run.txt
As is hopefully clear, both the CPU and GPU runs use the same input file (input_file.namd). They will each output to a separate log file (namd_output__cpu_run.txt and namd_output__gpu_run.txt). The final line of each section uses the grep utility to print the performance of each run in days per nanosecond (where a lower number indicates better performance).
If you’d like to visualize your results, you will need an SSH client which properly forwards your X-session. You are welcome to contact us if you’re uncertain of this step. Once that’s done, the VMD visualization tool can be run:
module load vmd vmd
Ready to try GPUs?
Once properly configured (which we’ve already done for you), running NAMD on a GPU cluster isn’t much more difficult than running it on your own workstation. This makes it easy to compare NAMD simulations running on CPUs and GPUs. If you’d like to give it a try, contact one of our experts or sign up for a GPU Test Drive today!
Citations for NAMD:
“NAMD was developed by the Theoretical and Computational Biophysics Group in the Beckman Institute for Advanced Science and Technology at the University of Illinois at Urbana-Champaign.”
James C. Phillips, Rosemary Braun, Wei Wang, James Gumbart, Emad Tajkhorshid, Elizabeth Villa, Christophe Chipot, Robert D. Skeel, Laxmikant Kale, and Klaus Schulten. Scalable molecular dynamics with NAMD. Journal of Computational Chemistry, 26:1781-1802, 2005. abstract, journal
https://www.ks.uiuc.edu/Research/namd/
Featured Illustration:
Molecular Dynamics of Viruses – Satellite Tobacco Mosaic Virus (STMV)
Citation for VMD:
Humphrey, W., Dalke, A. and Schulten, K., “VMD – Visual Molecular Dynamics” J. Molec. Graphics 1996, 14.1, 33-38
https://www.ks.uiuc.edu/Research/vmd/
